Mensaje Bioquímico XLV, 2021

Síntesis química de oligonucleótidos: Método de fosforamiditas

Instituto de Fisiología Celular, Circuito Exterior s/n, Ciudad Universitaria, Coyoacán, CDMX, México. CP 04510
Tel. +52 (55) 56 22 56 56, longay@ifc.unam.mx
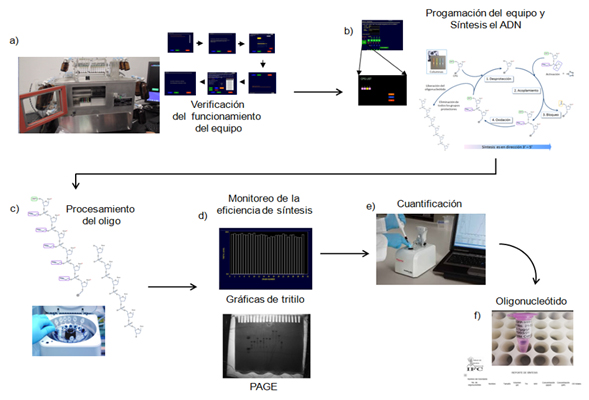
Los oligonucleótidos son cadenas de ácido nucléico relativamente cortas; van de unos cuantos nucleótidos hasta unos 200 nucleótidos los más largos. El uso de estas moléculas es de gran importancia en la biología molecular ya que se emplean en una gran variedad de técnicas como son el ADN recombinante, la mutagénesis dirigida, los microarreglos, la PCR, y la secuenciación, entre otras. Los oligonucleótidos generalmente se producen por síntesis química. Actualmente el método más utilizado es el de fosforamiditas en fase sólida, utilizando como soporte perlas de vidrio con poros de tamaño controlado (CGP). Este proceso utiliza nucleósidos modificados que tienen sus grupos reactivos protegidos, los cuales se activan de manera específica en el momento adecuado, lo que promueve la síntesis lineal, y evita la ramificación del oligonucleótido. La síntesis involucra la adición secuencial de los nucleósidos en dirección 3’-5’: el nucleósido es incorporado a la cadena creciente mediante un ciclo de síntesis que consta de cuatro pasos: desprotección, acoplamiento, bloqueo y oxidación. El ciclo se repite hasta obtener el oligonucleótido de la longitud deseada. Una vez que se completa la síntesis el oligonucleótido es liberado del soporte sólido y se eliminan todas las protecciones para obtener una molécula de ADN de cadena sencilla activa. Esta molécula se purifica y se concentra por precipitación alcohólica. El proceso de síntesis se lleva a cabo en sintetizadores automatizados con muy alta eficiencia, reproducibilidad y rendimiento. La calidad del oligonucleótido sintetizado se puede evaluar en geles desnaturalizantes de poliacrilamida. Los oligonucleótidos son estables por varios años si se almacenan a -20°C y libres de nucleasas.
Reacción en Cadena de la Polimerasa en Tiempo Real
Torre de Investigación, Facultad de Medicina, Ciudad Universitaria, Coyoacan, CDMX, México. CP 04510
Tel. +52 (55) 56 23 22 58, grodrig@unam.mx
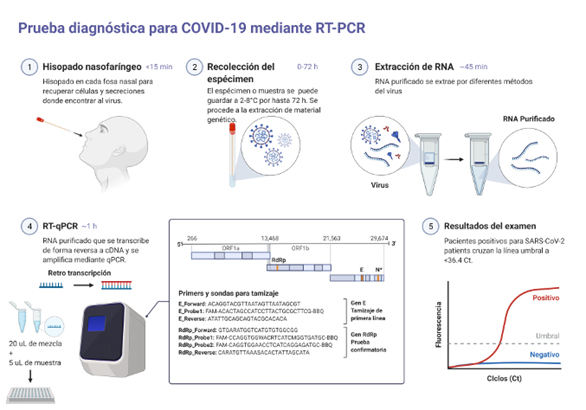
El desarrollo de la reacción en cadena de la polimerasa (PCR), realizada por Kary B. Mullis en 1983, revolucionó la biología molecular. Es una técnica para amplificar ácidos nucleicos, utilizando cebadores y una polimerasa termoestable. Este descubrimiento, acoplado a la química fluorescente y con el uso de la transcripción reversa, sentaron las bases para el desarrollo de la qPCR. El objetivo de la PCR en tiempo real es distinguir y cuantificar con precisión secuencias de ácidos nucleicos específicas en una muestra, monitoreando su amplificación a través de la detección de fluorescencia, ya que el aumento en la fluorescencia es proporcional a la acumulación del producto. Esta técnica tiene diversos campos de aplicación (ciencias básicas, biotecnología, medicina, etc.).
Secuenciación de ADN por el método de terminación de la cadena de Sanger
Instituto de Fisiología Celular, Circuito Exterior s/n, Ciudad Universitaria, Coyoacán, CDMX, México. CP 04510
Tel. +52 (55) 56 22 56 56, longay@ifc.unam.mx
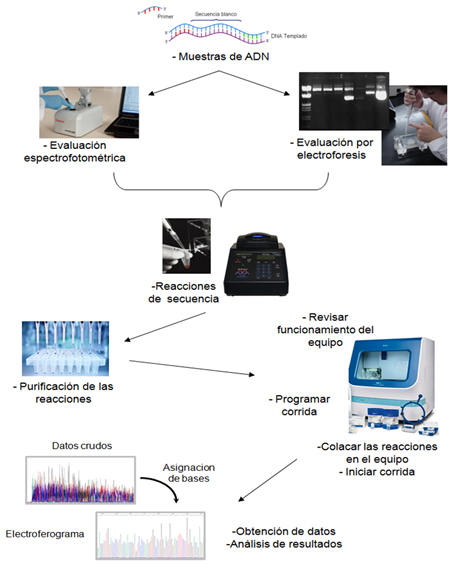
La secuenciación de ADN permite determinar el orden de los nucleótidos (A, G, C, T) en una molécula. El descubrimiento de la estructura de la molécula de ADN como una doble hélice de cadenas complementarias y la demostración de que esta molécula contiene la información genética, puso en evidencia la importancia de descifrar el orden de los nucleótidos en la cadena de ADN. La mayoría de las técnicas de secuenciación a baja escala que se usan actualmente se basan en el método de síntesis enzimática conocido como método Sanger o de terminación de la cadena. El principio de este método es la incorporación selectiva de dideoxinucleótidos trifosfato (ddNTPs), estos nucleótidos carecen del OH en el extremo 3’, por lo que si se incorporan durante la síntesis se impide la incorporación de un nuevo nucleótido y se termina la elongación de la cadena, de esta forma se generan fragmentos de distintos tamaños que terminan en todos los nucleótidos de la molécula de ADN, los cuales son separados por electroforesis para determinar la secuencia a partir del ddNTP incorporado y el tamaño de los fragmentos. Las técnicas actuales utilizan dideoxinucleotidos marcados con fluorocromos, para la identificación de los fragmentos. Se emplean ADN polimerasas termoestables que permiten hacer la reacción de secuencia por termo-ciclaje. Los productos de la reacción se separan por electroforesis capilar en secuenciadores automáticos. Estas modificaciones han incrementado la velocidad del proceso, la longitud de lectura y reducido significativamente los tiempos de corrida. Este tipo de secuenciación tiene alta eficiencia, es muy precisa y reproducible. Este método es útil para la secuenciación de genes o fragmentos individuales de ADN y en proyectos de secuenciación a baja escala que involucran un número reducido de fragmentos.
Purificación de proteínas
Instituto de Biotecnología, UNAM Av. Universidad # 2001 Col. Chamilpa, Cuernavaca, Morelos, México. CP 62210
Tel. +52 (55) 56 22 86 02, claudiar@comunidad.unam.mx
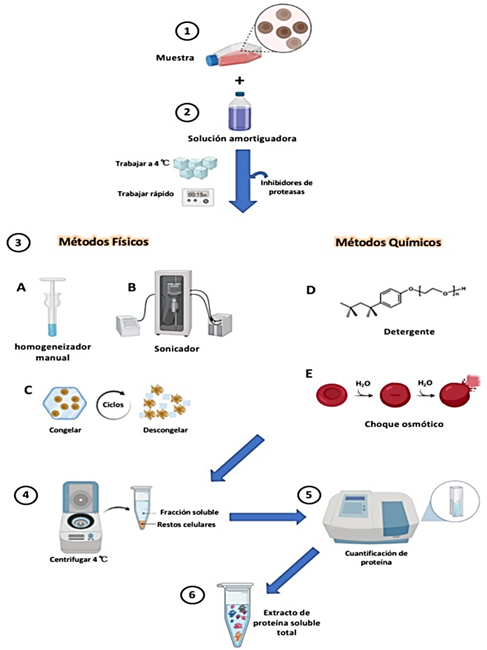
El desarrollo e innovación de técnicas que permitan un estudio cada vez más detallado de las proteínas, diverge en dos direcciones, hacia el conocimiento básico para conocer su papel en diferentes procesos celulares, y en otro sentido sus aplicaciones en las áreas de la biomedicina, biotecnología y nanotecnología. Las técnicas de purificación de proteínas juegan un papel fundamental en estas investigaciones, tomando como base el conocimiento de las propiedades de estas biomoléculas para determinar el protocolo de purificación. Un protocolo para purificar una proteína se inicia seleccionando la muestra biológica, el método y la condición para lisar las células con la finalidad de exponer a las proteínas, inevitablemente obteniendo una mezcla de estas junto con todos los componentes de la célula, biomoléculas, metabolitos y fragmentos de estructuras celulares. A partir de este momento, es imperante mantener a la proteína de interés en condiciones óptimas que le permitan preservar su función y estructura. La gama de métodos para la purificación de proteínas la podemos enlistar con base a estas propiedades; los métodos más comunes están basados en: purificación por peso molecular (cromatografía de filtración en gel), por carga (cromatografía de intercambio iónico), por hidrofobicidad (precipitación por sales o solventes, cromatografía de interacción hidrofóbica o fase reversa), por afinidad a un ligando (cromatografía de afinidad). La validación de cada técnica en un protocolo de purificación se basa en el uso de algunas de las variantes de la electroforesis en gel de poliacrilamida y cuantificación de proteínas. El éxito de una purificación de proteínas consiste en la paciencia, orden, y conocimiento previo de la proteína de interés, del fundamento de cada técnica, análisis detallado de cada paso, y sobre todo persistencia.
Ingeniería de proteínas
Instituto de Biotecnología, UNAM Av. Universidad # 2001 Col. Chamilpa, Cuernavaca, Morelos, México. CP 62210
Tel. +52 (55) 56 22 86 02, claudiar@comunidad.unam.mx
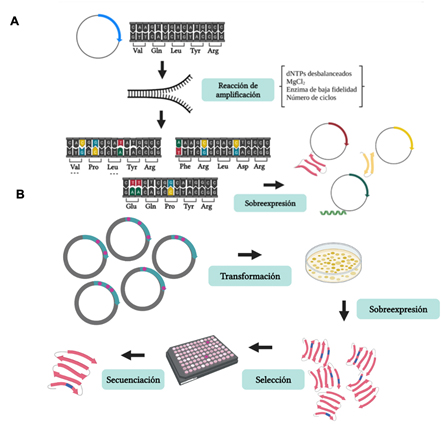
El conocimiento de los procesos evolutivos de la vida en la Tierra, ha dado la pauta en la innovación de llevar algunos de estos procesos al laboratorio para beneficio de la humanidad, como es la replicación de ADN y la síntesis de proteínas. El ADN contiene la información necesaria para la producción de una proteína. En el siglo pasado, se comenzaron a sentar las bases teóricas y experimentales para replicar ADN en el laboratorio, generando una de las técnicas experimentales fundamentales en la biología molecular, la reacción en cadena de la polimerasa (“Polymerase Chain Reaction”, PCR). A partir de la cual se comenzaron a implementar otras técnicas como la mutagénesis y la evolución dirigida, que finalmente forman parte de la Ingeniería de proteínas. La Ingeniería de proteínas consiste en modificar una proteína en un solo residuo de aminoácido, en una región, eliminando o insertando un fragmento de otra proteína, o fusionando dos proteínas, todo esto a partir de modificar la secuencia de ADN, con la finalidad de obtener una proteína con mejores características que la proteína inicial.
Producción de anticuerpos monoclonales murinos
Instituto de Diagnóstico y Referencia Epidemiológico “Dr. Manuel Martínez Báez” (InDRE),
Tel. +52 (55) 50 62 16 00 Ext. 59349, 59336, yolanda.medina@salud.gob.mx
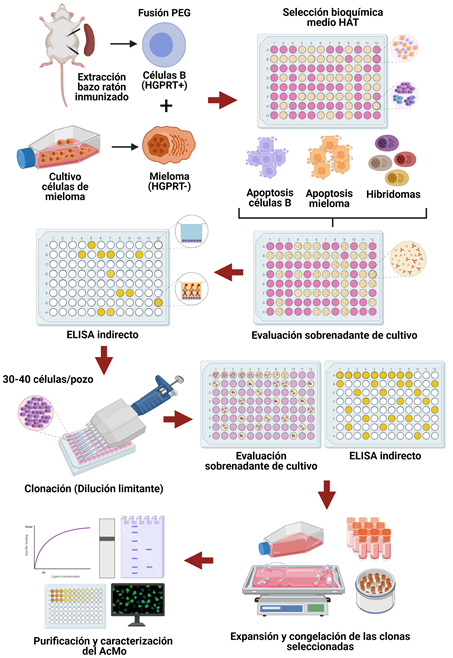
Los anticuerpos monoclonales son producidos por una sola clona de linfocitos B y tienen la característica principal de reconocer específicamente un solo epítopo en el antígeno. Actualmente, son ampliamente utilizados en la investigación, diagnóstico y tratamiento de enfermedades infecciosas, autoinmunes y cáncer. Los métodos de producción de anticuerpos monoclonales son diversos, entre ellos se encuentran; la inmortalización de linfocitos B, phage display, cultivo de células CHO e insectos, sin embargo, el método más utilizado es el descrito por Köhler y Milstein en 1975 que consiste en la producción de células híbridas. Los hibridomas son generados mediante la fusión de células del bazo de un ratón inmunizado con el antígeno de interés y las células de un plasmocitoma. Posterior a la fusión, se realiza una selección bioquímica en medio HAT (Hipoxantina-Timidina-Aminopterina), que permitirá la supervivencia y selección especifica de hibridomas. A continuación, se evalúa por ELISA indirecto la presencia de anticuerpos que reconocen el antígeno de interés a partir del sobrenadante de cultivo de los hibridomas, con la finalidad de identificar pozos en la placa de cultivo con híbridos productores. Finalmente, se realizan al menos dos ciclos de clonaciones con el objetivo de aislar un solo clon celular, el cual será expandido hasta obtener suficiente sobrenadante de cultivo para realizar la caracterización completa del anticuerpo producido: clase y subclase; especificidad, actividad biológica y aplicación final.
Método de inducción, aislamiento y selección de proteínas HRP de bacterias fitopatógenas, una fuente potencial de inductores de resistencia vegetal
Centro de Investigación y Asistencia en Tecnología y Diseño del Estado de Jalisco, A.C., Camino Arenero 1227, Col. El Bajío del Arenal, Jalisco, México. CP. 45019
Tel. +52 (33) 33 45 52 00, grincon@ciatej.mx
 Resumen
Resumen
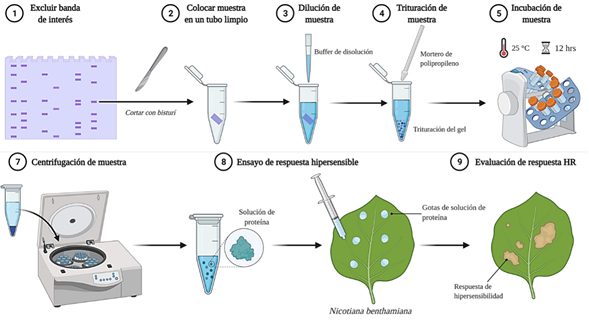
Xanthomonas vesicatoria es una bacteria Gram negativa y patógena de una gran variedad de especies vegetales, entre ellas especies de importancia económica para México como chile (Capsicum annuum) y jitomate (Solanum licopersicum). El grupo de genes hrp es conservado entre especies de Xanthomonas, dentro de este grupo de genes está el que codifica para el sistema de secreción tipo III y efectores patógenos, componentes clave en los procesos de infección de las células bacterianas a su planta huésped. Particularmente, proteínas HRP de Xanthomonas son producidas in situ en su planta hospedera durante los procesos de infección o in vitro en medios mínimos de crecimiento que imitan condiciones similares a su hospedero. El objetivo de este trabajo fue establecer un método de inducción, aislamiento y selección de proteínas HRP de bacterias fitopatógenas. Para ello, se diseñaron medios de inducción de proteínas HRP para Xanthomonas que comprenden tejido vegetal liofilizado de plantas hospederas como única fuente de carbono y nutrientes. Xanthomonas vesicatoria cepa BV801 (XAN801) fue capaz de crecer en los medios de inducción denominados AWSL y AWCA, conteniendo al menos 0.1% de tejido liofilizado de S. licopersicum y C. annuum respectivamente. El perfil de proteínas de XAN801 se resolvió por SDS-PAGE. Se demostró la inducción de bandas de proteínas en XAN801 cultivada en medios AWSL y AWCA. El aislamiento de bandas de proteínas inducidas se realizó por exclusión de geles de poliacrilamida después de su separación por SDS-PAGE. Las bandas de proteínas inducidas mantuvieron actividad biológica de respuesta hipersensible en hojas de tabaco (Nicotiana benthamiana). Los resultados de este trabajo son clave en la búsqueda y selección de proteínas HRP de bacterias fitopatógenas con capacidad de activar el sistema de defensa vegetal, lo cual puede conducir al fenómeno de resistencia fisiológica a fitopatógenos en plantas.
Aislamiento, purificación y cultivo de hepatocitos de rata
Departamento de Bioquímica, Edificio D, Facultad de Medicina, Circuito Escolar, Ciudad Universitaria, Coyoacan, CDMX, México. CP. 04510 Tel. +52 (55) 56 23 25 10, vilchisl@bq.unam.mx
 Resumen
Resumen
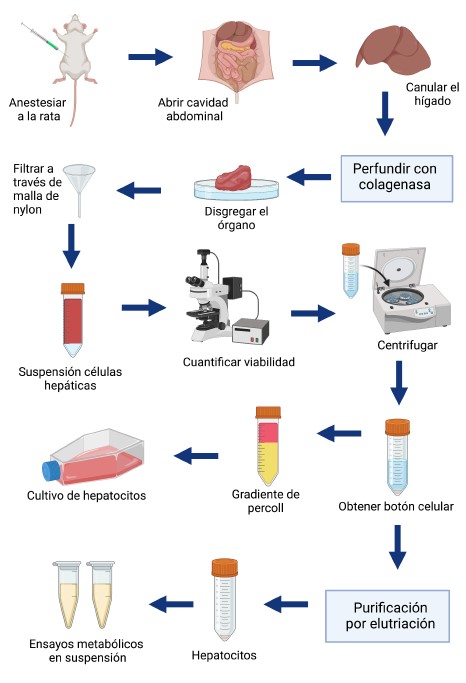
El órgano implicado en el metabolismo y la toxicidad de los xenobióticos es el hígado. Las principales células que forman parte de este órgano son los hepatocitos que se denominan genéricamente como células parenquimatosas y desempeñan las funciones metabólicas de este órgano. Por tanto, los hepatocitos
aislados de varias especies, incluida la humana y sus cultivos, constituyen modelos in vitro atractivos para la investigación básica de la función hepática, la fisiopatología, farmacología, y otros temas relacionados con el hígado. El método para el aislamiento de hepatocitos intactos se basa en la perfusión del hígado con colagenasa y fue introducido por primera vez por Berry y Friend en 1969, y desde entonces, ha sufrido modificaciones. Esencialmente, los hepatocitos se disocian del hígado de ratas adultas anestesiadas, por una perfusión de colagenasa a través de la vena porta. Las células aisladas se filtran a través de una malla de nylon para eliminar los restos de tejido no deseados y se obtiene una suspensión enriquecida de células parenquimatosas que se puede purificar mediante el método de elutriación para obtener hepatocitos puros. Los hepatocitos se pueden utilizar inmediatamente para realizar ensayos metabólicos en suspensión o se pueden cultivar en placas y utilizarlos por un periodo de 3 a 5 días.
Historia, fundamentos y métodos de la electroforesis de
proteínas en geles de poliacrilamida
Departamento de Biología, Facultad de Química, Circuito Exterior s/n, Ciudad Universitaria, Coyoacan, CDMX, México. CP. 04510 Tel. +52 (55) 56 22 38 99 Ext. 44449 y 44450, jjgartre@unam.mx
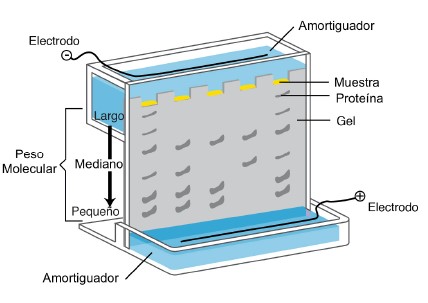 Resumen
Resumen
La electroforesis es una de las técnicas más poderosas en la bioquímica y la biología molecular modernas, dado que permite separar macromoléculas para estudiarlas de manera aislada y definir sus propiedades, funciones y mecanismos en las importantes reacciones biológicas de los seres vivos. En este artículo nos concentraremos en la electroforesis de proteínas. Las funciones biológicas, aunque codificadas en el ADN o ARN en general las llevan a cabo las proteínas en todos los seres vivos, ya sea en forma de enzimas, receptores, transportadores, bombas primarias y secundarias, proteínas estructurales, etc. Es por ello que la electroforesis de proteínas es una técnica tan poderosa, no sólo permite analizar la pureza de una muestra, sino que ayuda a determinar interacciones con otras proteínas, sustratos, ligandos, etc, y además ayuda a determinar los niveles de expresión de las proteínas dentro de las células, lo cual permite definir la relevancia de la función de una proteína dentro de cualquier ser vivo. Por todas estas razones, la electroforesis de proteínas se revisará en su historia y fundamentos, así como los protocolos más comunes para su uso en los laboratorios ya sean de investigación, biomédicos, farmacéuticos, biotecnológicos o clínicos.
Expresión de proteínas recombinantes en un sistema
heterólogo
Laboratorio de péptidos y proteínas, Torre de Investigación, Departamento de Bioquímica, Facultad de Medicina, UNAM; , Circuito Escolar, Ciudad Universitaria, Coyoacan, CDMX, México. CP. 04510 Tel. +52 (55) 56 23 21 33, ghernandez@bq.unam.mx
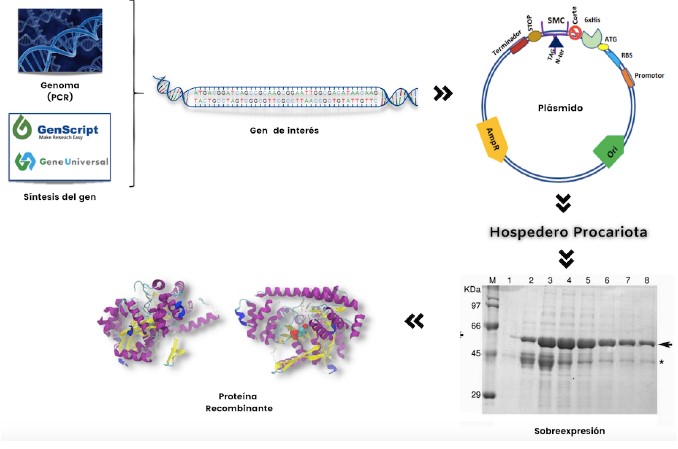 Resumen
Resumen
Los sistemas de expresión en organismos procariontes y eucariontes se utilizan comúnmente para la sobreexpresión de proteínas recombinantes solubles. La elección de un sistema de producción adecuado es la base para lograr un buen rendimiento en la purificación de la proteína recombinante de interés. Aspectos como: la optimización del uso de codones, la elección de la cepa hospedera, el vector de expresión, la etiqueta o proteína de fusión a utilizar para la purificación, la composición del medio de cultivo, las condiciones de inducción, entre otros factores, son la clave para el resultado final. El nivel de expresión del gen blanco, así como los procesamientos post-traduccionales de la proteína recombinante sintetizada son algunos de los principales retos a los que se enfrentan la mayoría de los estudios realizados. Uno de los sistemas de sobreexpresión más utilizados sin duda es el procariota, en los que se incluyen principalmente a Escherichia coli. Esto debido a la facilidad de su manipulación genética, crecimiento celular, gran cantidad de biomasa, bajo costo, y la gran cantidad de cepas hospederas disponibles. De tal forma, que E. coli ha superado la barrera de realizar algunas modificaciones post-traduccionales que requieren algunas proteínas de origen eucariota para producción en este sistema. En este trabajo se discuten algunas estrategias que han ayudado a la obtención de proteínas recombinantes activas y funcionales en un sistema comercial de sobreexpresión como el BL21 y así mismo, se sugieren algunos protocolos para determinar las mejores condiciones de sobreexpresión de las proteínas recombinantes de interés.
Aplicaciones de simulaciones de dinámica molecular en
proteínas
Laboratorio de Biosensores y Modelaje Molecular, Torre de Investigación, Departamento de Bioquímica, Facultad de Medicina, UNAM; Circuito Escolar, Ciudad Universitaria, Coyoacan, CDMX, México. CP. 04510 Tel. +52 (55) 56 23 22 54, martin@bq.unam.mx
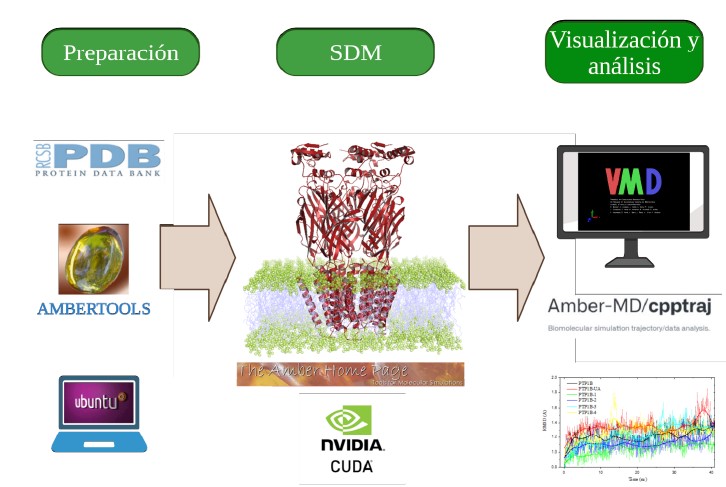 Resumen
Resumen
Las Simulaciones de Dinámica Molecular de proteínas y sus complejos representan actualmente una herramienta eficaz para describir a nivel atómico sistemas biológicos complejos y poder responder preguntas de investigación asociadas a estos sistemas. Esta herramienta computacional es un complemento idóneo a la parte experimental, con diversas aplicaciones para ser utilizada tales como el refinamiento de modelos estructurales, cálculos de parámetros energéticos de unión (proteína-ligando), estudio de plegamiento de proteínas, mecanismos de catálisis enzimática entre los más importantes. Las Simulaciones de Dinámica Molecular sea han visto beneficiadas del avance tecnológico de los sistemas informáticos, tanto a nivel hardware como a nivel software. Lo anterior, ha permitido tener una gran oferta de software y disminuir el tiempo de cómputo. En este protocolo, proporcionamos conceptos básicos, fundamentos de la técnica, aplicaciones de las Simulaciones de Dinámica Molecular, softwares disponibles para realizarlas y analizarlas; así como un protocolo general para un sistema proteínaligando.
Uso del sistema CRISPR-Cas9 para la edición de genes en organismos modelo y líneas celulares
Departamento de Biología Celular y Desarrollo, Instituto de Fisiología Celular, UNAM; Circuito Escolar, Ciudad Universitaria, Coyoacan, CDMX, México. CP. 04510 Tel. +52 (55) 56 22 56 09, rnavarro@ifc.unam.mx
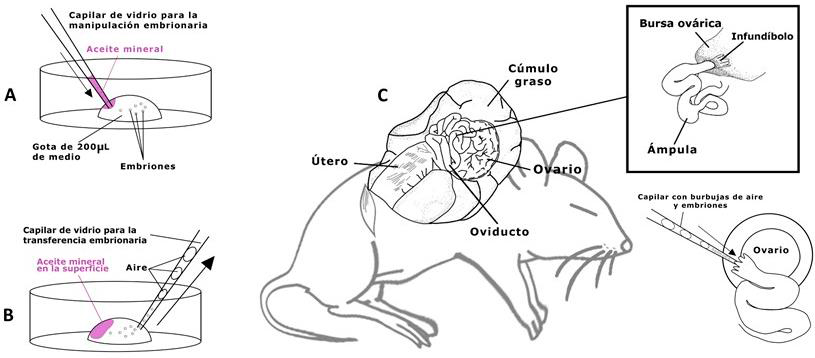 Resumen
Resumen
El sistema CRISPR-Cas9 es una poderosa tecnología que permite editar el genoma eucarionte de manera rápida, precisa y eficiente. Esta herramienta ha revolucionado la manera de modificar el genoma de distintos organismos, desde microorganismos hasta mamíferos. Mediante el uso de CRISPR-Cas no es solo posible introducir mutaciones para estudiar la ausencia de un determinado gene, sino que también nos permite editar el genoma para introducir marcadores fluorescentes o incluso editar el epigenoma. Los protocolos de CRISPR-Cas9 se basan en introducir a las células tanto a la proteína Cas9 como a RNAs guías que dirigen al sistema junto con templados de homología. En este capítulo presentamos protocolos detallados para aplicar esta tecnología de edición genómica en distintos organismos modelo como el nematodo Caenorhabditis elegans, líneas celulares de la mosca de la fruta Drosophila melanogaster, el pez cebra Danio rerio y ovocitos de ratón. Esperamos que este capítulo permita a distintos grupos de investigación aplicar esta poderosa tecnología en sus modelos experimentales